 |
|
|
|
Sun, 26 Jul, 2026
|

|

|
Bioinformatics or computational biology is the use of techniques from applied mathematics, informatics, statistics, and computer science to solve biological problems. Research in computational biology often overlaps with systems biology. Major research efforts in the field include sequence alignment, gene finding, genome assembly, protein structure alignment, protein structure prediction, prediction of gene expression and protein-protein interactions, and the modeling of evolution.The terms bioinformatics and computational biology are often used interchangeably, although the latter typically focuses on algorithm development and specific computational methods. (In the biology-mathematics-computer science triad, bioinformatics will intimately involve all three components while computational biology will focus on biology and mathematics.) Due to interest from computer scientists and mathematicians and the popularity of computational techniques in the field of genomics, it is commonly referred to as computational biology; a more accurate term is computational genomics. There are also lesser known but equally important areas of computational biochemistry and computational biophysics, that are also a part of computational biology. (For working definitions of Bioinformatics and Computational Biology used by National Institutes of Health please see this link.) A common thread in projects in bioinformatics and computational genomics is the use of mathematical tools to extract useful information from noisy data produced by high-throughput biological techniques. (The field of data mining overlaps with computational biology in this regard.) Representative problems in computational biology include the assembly of high-quality DNA sequences from fragmentary "shotgun" DNA sequencing, and the prediction of gene regulation with data from mRNA microarrays or mass spectrometry.
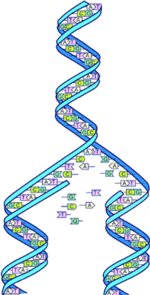 Making sense of the huge amounts of DNA data (pictured) produced by gene sequencing projects is just one of the tasks faced by bioinformatics.
 Jump to Page Contents Jump to Page Contents
|
|

Pay as you go
No monthly charges. Access for the price of a phone call
Go>
Unmetered
Flat rate dialup access from only £4.99 a month Go>
Broadband
Surf faster from just £13.99 a month Go> |
Save Even More
Combine your phone and internet, and save on your phone calls
More Info> |
This weeks hot offer
 24: Series 5
24: Series 5
In association with Amazon.co.uk £26.97 |
|
Contents
Major Research Areas
Software tools
Notes & references
Major Research Areas - Contents
Sequence analysis
Since the Phage Φ-X174 was sequenced in 1977, the DNA sequences of more and more organisms have been decoded and stored in electronic databases. This data is analyzed to determine genes that code for proteins, as well as regulatory sequences. A comparison of genes within a species or between different species can show similarities between protein functions, or relations between species (the use of molecular systematics to construct phylogenetic trees). With the growing amount of data, it long ago became impractical to analyze DNA sequences manually. Today, computer programs are used to search the genome of thousands of organisms, containing billions of nucleotides. These programs can compensate for mutations (exchanged, deleted or inserted bases) in the DNA sequence, in order to identify sequences that are related, but not identical. A variant of this sequence alignment is used in the sequencing process itself. The so-called shotgun sequencing technique (which was used, for example, by The Institute for Genomic Research to sequence the first bacterial genome, Haemophilus influenza) does not give a sequential list of nucleotides, but instead the sequences of thousands of small DNA fragments (each about 600-800 nucleotides long). The ends of these fragments overlap and, when aligned in the right way, make up the complete genome. Shotgun sequencing yields sequence data quickly, but the task of assembling the fragments can be quite complicated for larger genomes. In the case of the Human Genome Project, it took several months of CPU time (on a circa-2000 vintage DEC Alpha computer) to assemble the fragments. Shotgun sequencing is the method of choice for virtually all genomes sequenced today, and genome assembly algorithms are a critical area of bioinformatics research.Another aspect of bioinformatics in sequence analysis is the automatic search for genes and regulatory sequences within a genome. Not all of the nucleotides within a genome are genes. Within the genome of higher organisms, large parts of the DNA do not serve any obvious purpose. This so-called junk DNA may, however, contain unrecognized functional elements. Bioinformatics helps to bridge the gap between genome and proteome projects, for example in the use of DNA sequence for protein identification.Genome annotation
In the context of genomics, annotation is the process of marking the genes and other biological features in a DNA sequence. The first genome annotation software system was designed in 1995 by Owen White, who was part of the team that sequenced and analyzed the first genome of a free-living organism to be decoded, the bacterium Haemophilus influenzae. Dr. White built a software system to find the genes (places in the DNA sequence that encode a protein), the transfer RNA, and other features, and to make initial assignments of function to those genes. Most current genome annotation systems work similarly, but the programs available for analysis of genomic DNA are constantly changing and improving.
Computational evolutionary biology
Evolutionary biology is the study of the origin and descent of species, as well as their change over time. Informatics has assisted evolutionary biologists in several key ways; it has enabled researchers to:
- trace the evolution of a large number of organisms by measuring changes in their DNA, rather than through physical taxonomy or physiological observations alone,
- more recently, compare entire genomes, which permits the study of more complex evolutionary events, such as gene duplication, lateral gene transfer, and the prediction of bacterial speciation factors,
- build complex computational models of populations to predict the outcome of the system over time
- track and share information on an increasingly large number of species and organisms
Future work endeavours to reconstruct the now more complex tree of life.The area of research within computer science that uses genetic algorithms is sometimes confused with computational evolutionary biology. Work in this area involves using specialized computer software to improve equations, algorithms, or integrated circuit designs. It is inspired by evolutionary principles such as replication, diversification through recombination or mutation, fitness, survival through selection or culling, and iteration, collectively called a Darwinian machine or Darwinian ratchet.
Measuring biodiversity
Biodiversity of an ecosystem might be defined as the total genomic complement of a particular environment, from all of the species present, whether it is a biofilm in an abandoned mine, a drop of sea water, a scoop of soil, or the entire biosphere of the planet Earth. Databases are used to collect the species names, descriptions, distributions, genetic information, status and size of populations, habitat needs, and how each organism interacts with other species. Specialized software programs are used to find, visualize, and analyze the information, and most importantly, communicate it to other people. Computer simulations model such things as population dynamics, or calculate the cumulative genetic health of a breeding pool (in agriculture) or endangered population (in conservation). One very exciting potential of this field is that entire DNA sequences, or genomes of endangered species can be preserved, allowing the results of Nature's genetic experiment to be remembered in silico, and possibly reused in the future, even if that species is eventually lost.Important Projects: Species 2000 project.
Gene expression analysis
The expression of many genes can be determined by measuring mRNA levels with multiple techniques including microarrays, expressed cDNA sequence tag (EST) sequencing, serial analysis of gene expression (SAGE) tag sequencing, massively parallel signature sequencing (MPSS), or various applications of multiplexed in-situ hybridization. All of these techniques are extremely noise-prone and/or subject to bias in the biological measurement, and a major research area in computational biology involves developing statistical tools to separate signal from noise in high-throughput gene expression (HT) studies. HT studies are often used to determine the genes implicated in a disorder: one might compare microarray data from cancerous epithelial cells to data from non-cancerous cells to determine the proteins that are up-regulated and down-regulated in cancer cells.
Regulation analysis
Regulation is the complex orchestra of events starting with an extra-cellular signal and ultimately leading to the increase or decrease in the activity of one or more protein molecules. Bioinformatics techniques have been applied to explore various steps in this process. For example, promoter analysis involves the elucidation and study of sequence motifs in the genomic region surround the coding region of a gene. These motifs influence the extent to which that region is transcribed into mRNA. Expression data can be used to infer gene regulation: one might compare microarray data from a wide variety of states of an organism to form hypotheses about the genes involved in each state. In a single-cell organism, one might compare stages of the cell cycle, along with various stress conditions (heat shock, starvation, etc.). One can then apply clustering algorithms to that expression data to determine which genes are co-expressed. Further analysis could take a variety of directions: one 2004 study analyzed the promoter sequences of co-expressed (clustered together) genes to find common regulatory elements and used machine learning techniques to identify the promoter elements involved in regulating each cluster.
Protein expression analysis
Protein microarrays and high throughput (HT) mass spectrometry (MS) can provide a snapshot of the proteins present in a biological sample. Bioinformatics is very much involved in making sense of protein microarray and HT MS data; the former involves a number of the same problems involve in examining microarrays targeted at mRNA, the latter involves the problem of matching large amounts of mass data against predicted masses from protein sequence databases, and the complicated statistical analysis of samples where multiple, but incomplete, peptides from each protein are detected.
Analysis of mutations in cancer
Massive sequencing efforts are currently underway to identify point mutations in a variety of genes in cancer. The sheer volume of data produced requires automated systems to read sequence data, and to compare the sequencing results to the known sequence of the human genome, including known germline polymorphisms.
Oligonucleotide microarrays, including comparative genomic hybridization and single nucleotide polymorphism arrays, able to probe simultaneously up to several hundred thousand sites throughout the genome are being used to identify chromosomal gains and losses in cancer. Hidden Markov model and change-point analysis methods are being developed to infer real copy number changes from often noisy data. Further informatics approaches are being developed to understand the implications of lesions found to be recurrent across many tumors.Some modern tools (e.g. Quantum 3.1 ) provide tool for changing the protein sequence at specific sites through alterations to its amino acids and predict changes in the bioactivity after mutations.
Structure prediction
Protein structure prediction is another important application of bioinformatics. The amino acid sequence of a protein, the so-called primary structure, can be easily determined from the sequence on the gene that codes for it. In the vast majority of cases, this primary structure uniquely determine a structure in its native environment. (Of course, there are exceptions, such as the bovine spongiform encephalopathy - aka Mad Cow Disease - prion.) Knowledge of this structure is vital in understanding the function of the protein. For lack of better terms, structural information are usually classified as one of secondary, tertiary and quaternary structures. A viable general solution to such predictions remains an open problem. As of now, most efforts have been directed towards heuristics that work most of the time.One of the key ideas in bioinformatics research is the notion of homology. In the genomic branch of bioinformatics, homology is used to predict the function of a gene: if the sequence of gene A, whose function is known, is homologous to the sequence of gene B, whose function is unknown, one could infer that B may share A's function. In the structural branch of bioinformatics homology is used to determine which parts of the protein are important in structure formation and interaction with other proteins. In a technique called homology modelling, this information is used to predict the structure of a protein once the structure of a homologous protein is known. This currently remains the only way to predict protein structures reliably.One example of this is the similar protein homology between hemoglobin in humans and the hemoglobin in legumes ( leghemoglobin). Both serve the same purpose of transporting oxygen in both organisms. Though both of these proteins have completely different amino acid sequences, their protein structures are virtually identical, which reflects their near identical purposes.Other techniques for predicting protein structure include protein threading and de novo (from scratch) physics-based modeling.See also structural motif and structural domain.
Comparative genomics
The core of comparative genome analysis is the establishment of the correspondence between genes (orthology analysis) or other genomic features in different organisms. It is these intergenomic maps that make it possible to trace the evolutionary processes responsible for the divergence of two genomes. A multitude of evolutionary events acting at various organizational levels shape genome evolution. At the lowest level, point mutations affect individual nucleotides. At a higher level, large chromosomal segments undergo duplication, lateral transfer, inversion, transposition, deletion and insertion. Ultimately, whole genomes are involved in processes of hybridization, polyploidization and endosymbiosis, often leading to rapid speciation. The complexity of genome evolution poses many exciting challenges to developers of mathematical models and algorithms, who have recourse to a spectra of algorithmic, statistical and mathematical techniques, ranging from exact, heuristics, fixed parameter and approximation algorithms for problems based on parsimony models to Markov Chain Monte Carlo algorithms for Bayesian analysis of problems based on probabilistic models.Many of these studies are based on the homology detection and protein families computation.See also comparative genomics, bayesian network and protein family.
Modeling biological systems
Systems biology involves the use of computer simulations of cellular subsystems (such as the networks of metabolites and enzymes which comprise metabolism, signal transduction pathways and gene regulatory networks) to both analyze and visualize the complex connections of these cellular processes. Artificial life or virtual evolution attempts to understand evolutionary processes via the computer simulation of simple (artificial) life forms.
High-throughput image analysis
Computational technologies are also used to accelerate or fully automate the processing, quantification and analysis of large amounts of high-information-content Biomedical imagery. Modern image analysis systems augment the observers ability to make measurements from a large or complex set of images, by improving accuracy, objectivity, or speed. A fully developed analysis system may completely replace the observer. While these systems are not unique to biology related imagery, their application to biologic problems continue to provide unique challenges and solutions, placing several imagery application under the umbrella of Bioinformatics. These systems are in the process of becoming more important for both diagnostics and research. Some examples:
- high-throughput and high-fidelity quantification and sub-cellular localization ( high-content screening, cytohistopathology)
-
morphometrics are used to analyze pictures of embryos to track and to predict the fate of cell clusters during morphogenesis
- clinical image analysis and visualization
- determine the real-time air-flow patterns in breathing lungs of living individuals before and during challenge
- quantify occlusion size in real-time imagery from the development of and recovery during arterial injury
- making behavioural observations from extended video recordings of laboratory animals
- infrared measurements for metabolic activity determination
Software tools - Contents
The computational biology tool best-known among biologists is probably BLAST, an algorithm for searching large sequence (protein, DNA) databases. NCBI provides a popular implementation that searches their massive sequence databases. Bioinformatic meta search engines ( Entrez, Bioinformatic Harvester) help finding relevant information from several databases. There are also free Web-based software designed for structural bioinformatics such as [2] STING.Computer scripting languages such as Perl and Python are often used to interface with biological databases and parse output from bioinformatics programs. Communities of bioinformatics programmers have set up free/open source projects such as EMBOSS, Bioconductor, BioPerl, BioLinux, BioPython, BioRuby, and BioJava which develop and distribute shared programming tools and objects (as program modules) that make bioinformatics easier.An integrated software workbench consisting of many free/open source tools described above and many others is known as VigyaanCD. Taverna an open-source bioinformatics workbench that utilises a workflow model of experimental design. Taverna is included as part of the myGRID package of e-science software. [ http://www.q-pharm.com Quantum 3.1] is an example of the bioinformatics post- QSAR technology applying quantum and molecular physics instead of statistical methods.
Notes & references - Contents
- ^ Beer MA, Tavazoie S. " Predicting gene expression from sequence." In Cell. 2004 Apr 16;117(2):185-98.]
|
Change Text Size:
[A]
[default]
[A] |


 |
|
|
|
|